multi-sample scATAC-seq
Gali Bai
6/6/2021
Last updated: 2021-07-27
Checks: 7 0
Knit directory: MAESTRO_documentation/
This reproducible R Markdown analysis was created with workflowr (version 1.6.2). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20201223) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility.
The results in this page were generated with repository version 4a7b06f. See the Past versions tab to see a history of the changes made to the R Markdown and HTML files.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .RData
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: analysis/.DS_Store
Ignored: code/.DS_Store
Ignored: data/.DS_Store
Ignored: data/multi-scatac/.DS_Store
Ignored: data/multi-scrna/
Untracked files:
Untracked: .RDataTmp
Untracked: code/snRNA_genelength.R
Untracked: code/snRNA_genelength_TPM.R
Untracked: data/allen/
Untracked: data/multi-scatac/bed/
Untracked: data/multi-scatac/bigwig/GDF7_bed_added.png
Untracked: output/snRNA_TPM_VS_genelength.png
Untracked: output/snRNA_genelengthVSLognormUMI.png
Untracked: output/snRNA_genelengthVSUMI.png
Unstaged changes:
Deleted: MultiSample_scATACseq.Rproj
Deleted: atac_pbmc_500_nextgem.GIGGLE/0.peaks.bed
Deleted: atac_pbmc_500_nextgem.GIGGLE/0.peaks.bed.gz
Deleted: atac_pbmc_500_nextgem.GIGGLE/0.peaks.bed.result.xls
Modified: code/Basic_Operations.R
Modified: pbmc_1k_v3_8k_res.rds
Modified: pbmc_1k_v3_Monocyte_filtered.pdf
Modified: pbmc_1k_v3_Monocyte_top.pdf
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
There are no past versions. Publish this analysis with wflow_publish() to start tracking its development.
Introduction
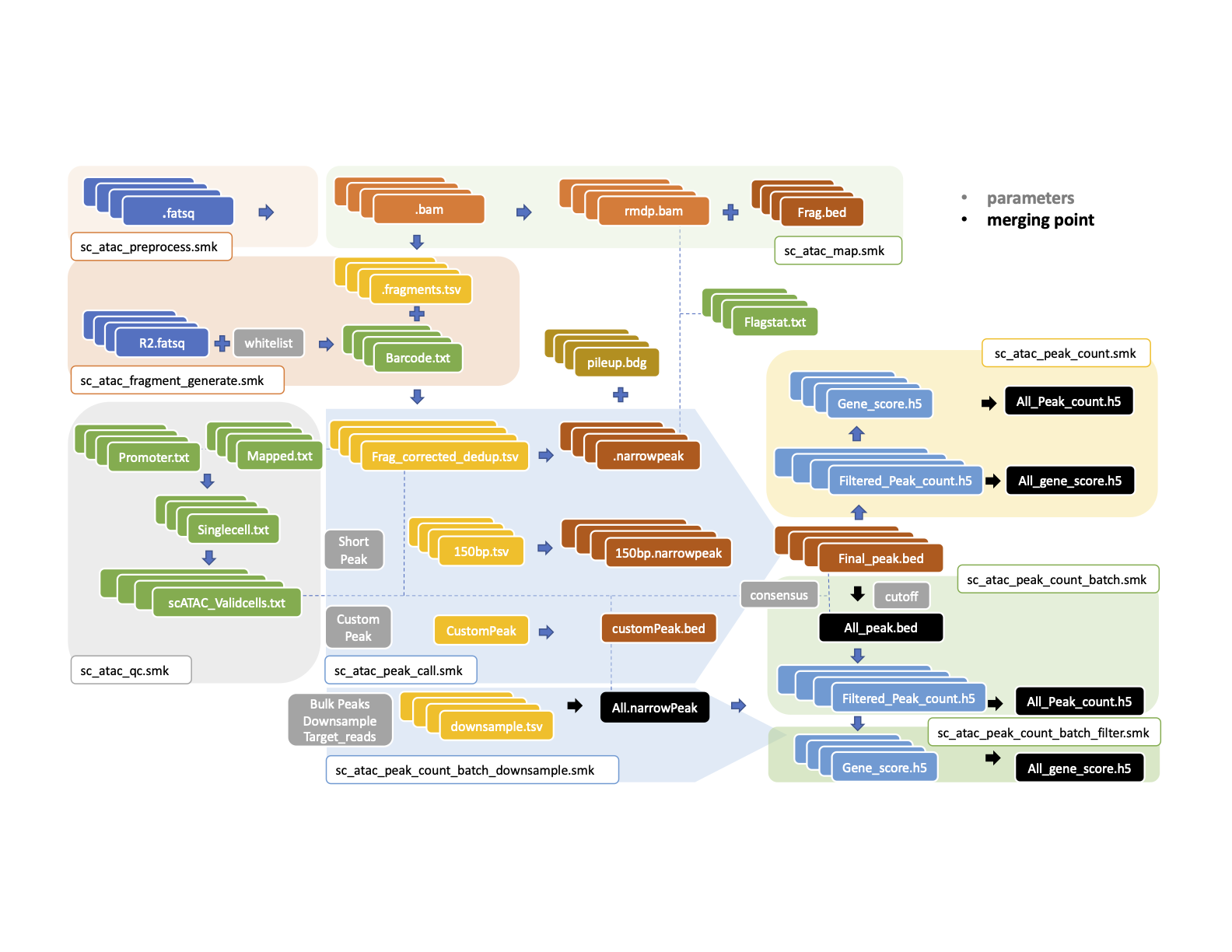
MAESTRO multi-sample scATAC-seq function is the extension of the scATAC-seq pipeline. By specifying different merging points, users can choose either to call bulk peaks or to call peaks by each sample individually. At the end of each workflow, users will obtain both individual peak/gene matrix and merged peak/gene matrix. Each merging point has been shown in the below flow chart. The downstream analysis will then be conducted based on the output matrix. After collecting clustering information, the multi-sample pipeline will also split the fragment file by samples and clusters. This step was developed to facilitate the comparison between samples within each cluster.
| Version | Author | Date |
|---|---|---|
| d88199f | baigal628 | 2020-12-29 |
Tutorial: PBMC scATAC-seq in time-Dependent Sampling Studies
Step0. Data Preparation
Create a list of accessions
$ touch SraAccList.txt
$ nano SraAccList.txtCopy the following accession numbers in SraAccList.txt:
$ mkdir data
$ cd data
$ touch SraAccList.txt
$ nano SraAccList.txt
#copy the following accession numbers
SRR11614703
SRR11614704
SRR11614705
SRR11614706
SRR11614707
SRR11614708#Install sratoolkit through conda
$ conda install -c bioconda sra-tools
#Split .sra to .fastq.gz
$ cat SraAccList.txt | while read i
do
time fastq-dump --gzip -split-files ${i}
echo "** ${i}.sra to fastq done **"
done
#Rename to 10X format
$ cat SraAccList.txt | while read i
do
mv ${i}_1*.gz ${i}_S1_L001_R1_001.fastq.gz
mv ${i}_2*.gz ${i}_S1_L001_R3_001.fastq.gz
mv ${i}_3*.gz ${i}_S1_L001_R2_001.fastq.gz
done
#I renamed each file based on sample information (Optional)
$ mv SRR11614703_S1_L001_R1_001.fastq.gz CLL0_S1_L001_R1_001.fastq.gzStep 1. Configure the MAESTRO workflow
Initialize the MAESTRO scATAC-seq workflow using MAESTRO scATAC-init command. This will install a Snakefile and a config file in this directory. Here we take the 10X PBMC and CLL data as an example. Considering MAESTRO provides built-in immune cell markers, it’s recommended to choose the RP-based cell-type annotation strategy. If the data is not immune-related, users can choose to provide their own marker gene list, or choose to annotate cell types through the peak-based method (see the following argument description for more details), or they can directly skip the annotation step by not setting --annotation.
$ MAESTRO scatac-init --batch --bulk_peaks --downsample \
--target_reads 5000000 --input_path /single-cell/MAESTRO/test/tutorial/data/time_data \
--gzip --platform 10x-genomics --format fastq --species GRCh38 \
--deduplication cell-level --mapping chromap \
--giggleannotation /single-cell/annotations/giggle.all/ \
--fasta /single-cell/references/Refdata_scATAC_MAESTRO_GRCh38_1.1.0/GRCh38_genome.fa \
--index /single-cell/references/chromap/GRCh38_chromap.index \
--whitelist /single-cell/references/whitelist/737K-cratac-v1.txt \
--cores 16 --directory multi-scatac-chromap \
--annotation --method RP-based --signature human.immune.CIBERSORT \
--clusterpeak --shortpeak \
--rpmodel Enhanced \
--peak_cutoff 100 --count_cutoff 1000 --frip_cutoff 0.2 --cell_cutoff 500To get a full description of command-line options, please use the command
$ MAESTRO scatac-init -hHere we list all the arguments and their description.
multi-sample processing parameters:
| Arguments | Description |
|---|---|
--batch |
If set as true, multi-sample pipeline will be initiated. Users can either choose to go through consensus-peaks path or bulk-peaks path (illustrated in workflow above), which are mutually exclusive. A count matrix for each sample will be produced based on the merged peak set. |
--consensus_peaks |
If true, –batch needs to be also set as true. Users can define whether to merge consensus peaks from each sample. In this way, MAESTRO will call peaks on each sample individually first and merge consensus peaks from all samples. Users should also set number of cutoff_samples to define the consensus (i.e. peaks called in >= cutoff_samples). |
--cutoff_samples |
Minimum number of samples to present consensus peaks. The peaks present in at least cutoff_samples will be kept. |
--bulk_peaks |
For multi-samples from the same experiment, if set as true, peaks will be called after merging all bam file. Bulk_peaks and consensus_peaks are mutually exclusive. |
--downsample |
{True,False} For deeply sequenced samples, bam files can be downsampled to a certain number of reads (target_reads) to get peak set. Effective only when bulk_peaks is true. |
--target_reads |
Number of reads to be kept in downsampling. If the sample has fewer than the target_reads, the original number of reads will be kept. Effective only when down_sample is true. |
Input files arguments:
| Arguments | Description |
|---|---|
--input_path |
Path to the input fastq files. |
--gzip |
Set as True if the input files are gzipped. |
--species |
{GRCh38,GRCm38} Specify the genome assembly (GRCh38 for human and GRCm38 for mouse). DEFAULT: GRCh38. |
--platform |
{10x-genomics,sci-ATAC-seq} Platform of single cell ATAC-seq. DEFAULT: 10x-genomics. |
--format |
{fastq,bam,fragments} The format of input files. Users can start with sequencing fastq files, bam files with CB tag or fragments.tsv.gz file generated by CellRanger ATAC. If the platform is 10x-genomics, users can start with sequencing fastq files, bam files with CB tag or fragments.tsv.gz file generated by CellRanger ATAC. If the platform is sci-ATAC-seq, users can start with sequencing fastq files, bam files with CB tag. |
--mapping |
{chromap,minimap2} Choose the aligment tool for scATAC-seq from either chromap or minimap2. DEFAULT: chromap. |
--deduplication |
{cell-level,bulk-level} deduplication level: cell-level or bulk-level. |
Reference genome arguments:
| Arguments | Description |
|---|---|
--giggleannotation |
Path of the giggle annotation file required for regulator identification. Please download the annotation file from here and decompress it.. |
--fasta |
Genome fasta file for minimap2. Users can just download the fasta file for human and mouse from CistromDB and decompress them. For example, --fasta Refdata_scATAC_MAESTR O_GRCh38_1.1.0/GRCh38_genome.fa. |
--index |
Path of the reference index file for chromap. Users need to build the index file for the reference using command $ chromap -i -r ref.fa -o index. |
Barcode library arguments, only for the platform of ‘sci-ATAC-seq’:
| Arguments | Description |
|---|---|
--whitelist |
If the platform is ‘sci-ATAC-seq’ or ‘10x-genomics’, please specify the barcode library (whitelist) so that the pipeline can correct cell barcodes with 1 base mismatched. Otherwise, the pipeline will automatically output the barcodes with enough read count (>1000). The 10X Chromium whitelist file can be found inside the CellRanger-ATAC distribution. For example, in CellRanger-ATAC 1.1.0, the whitelist is ‘cellranger-atac-1.1.0/cellranger-atac-cs/1.1.0/lib/python/barcodes/737K-cratac-v1.txt’. |
Output arguments:
| Arguments | Description |
|---|---|
--cores |
The number of cores to use. DEFAULT: 8. |
--directory |
Path to the directory where the workflow shall be initialized and results shall be stored. DEFAULT: MAESTRO. |
Cell-type annotation arguments:
| Arguments | Description |
|---|---|
--annotation |
Whether or not to perform cell-type annotation. By default (not set), MAESTRO will skip the step of cell-type annotation. If set, please specify the method of cell-type annotation through --method. |
--method |
{RP-based,peak-based,both} Method to annotate cell types. MAESTRO provides two strategies to annotate cell types for scATAC-seq data. Users can choose from ‘RP-based’ and ‘peak-based’, or choose to run both of them. One is based on gene regulatory potential predicted by RP model. Another is based on the bulk chromatin accessibility data from Cistrome database. If ‘RP-based’ is set, MAESTRO performs the cell-type annotation using the gene regulatory potential to represent gene expression, and the logFC of gene regulatory potential between one cluster and all the other cells is used to calculate the gene signature scores. If ‘peak-based’ is set, MAESTRO utilizes GIGGLE to evaluate the enrichment of bulk chromatin accessibility peaks on cluster-specific peaks from scATAC-seq data, and then transfers the Cistrome cluster identity from the most enriched bulk chromatin accessibility data as the cell-type annotation for the scATAC-seq cluster. See the MAESTRO paper for more details. DEFAULT: RP-based. |
--signature |
Cell signature file used to annotate cell types (required when method is set as ‘RP-based’). MAESTRO provides several sets of built-in cell signatures. Users can choose from [‘human.immune.CIBERSORT’, ‘mouse.brain.ALLEN’, ‘mouse.all.facs.TabulaMuris’, ‘mouse.all.droplet.TabulaMuris’]. Custom cell signatures are also supported. In this situation, users need to provide the file location of cell signatures, and the signature file is tab-seperated without header. The first column is cell type, and the second column is signature gene. DEFAULT: human.immune.CIBERSORT. |
Customized peak arguments:
| Arguments | Description |
|---|---|
--custompeak |
Whether or not to provide custom peaks. If set, users need to provide the file location of peak file through --custompeak-file and then MAESTRO will merge the custom peak file and the peak file called from all fragments using MACS2. By default (not set), the pipeline will use the peaks called using MACS2. |
--custompeak-file |
If --custompeak is set, please provide the file location of custom peak file. The peak file is BED formatted with tab-separated. The first column is the chromosome, the second is chromStart, and the third is chromEnd. |
--shortpeak |
Whether or not to call peaks from short fragments (shorter than 150bp). If set, MAESTRO will merge the peaks called from all fragments and those called from short fragments, and then use the merged peak file for further analysis. If not (by default), the pipeline will only use peaks called from all fragments. |
--clusterpeak |
Whether or not to call peaks by cluster. If set, MAESTRO will split the bam file according to the clustering result, and then call peaks for each cluster. By default (not set), MAESTRO will skip this step. |
Gene score arguments:
| Arguments | Description |
|---|---|
--rpmodel |
{Simple,Enhanced} The RP model to use to calaculate gene score. For each gene, simple model sums over the impact of all regulatory elements within the up/dowm-stream of TSS. On the basis of simple model, enhanced model gives the regulatory elements within the exon region a higher weight, and also excludes the regulatory elements overlapped with another gene (the promoter and exon of a nearby gene). See the MAESTRO paper for more details. DEFAULT: Enhanced. |
--genedistance |
Gene score decay distance, could be optional from 1kb (promoter-based regulation) to 10kb (enhancer-based regulation). DEFAULT: 10000. |
Quality control arguments:
| Arguments | Description |
|---|---|
--peak-cutoff |
Minimum number of peaks included in each cell. DEFAULT: 100. |
--count-cutoff |
Cutoff for the number of count in each cell. DEFAULT: 1000. |
--frip-cutoff |
Cutoff for fraction of reads in promoter in each cell. DEFAULT: 0.2. |
--cell-cutoff |
Minimum number of cells covered by each peak. DEFAULT: 10. |
Step 2. Configure samples.json file
In the project directory, we will next run MAESTRO samples-init subcommand to configure samples.json file. The purpose of this step is let MAESTRO read the right format and path of input data.
$ cd multi-scatac-chromap/
$ MAESTRO samples-init --assay_type scatac --platform 10x-genomics --data_type fastq --data_dir /single-cell/MAESTRO/test/tutorial/data/time_dataTo get a full description of command-line options, please use the command
MAESTRO samples-init -hInput files arguments:
| Arguments | Description |
|---|---|
--assay_type |
{scrna,scatac} choose the type of assays. |
--platform |
{10x-genomics,sci-ATAC-seq,microfluidic} choose the type of sequencing platform. |
--data_type |
{fastq,fragment,bam} choose the type of input data. When using fastq format as input, MAESTRO can only recognize the 10X-like naming: [Sample Name]_S1_L00[Lane Number]_[Read Type]_001.fastq.gz. |
--data_dir |
the full path to the input file folder. |
Once configured, you will find a samples.json file in the current project directory:
$ cat samples.json
{
"CLL0": {
"R1": [
"/MAESTRO/test/tutorial/data/time_data/CLL0_S1_L001_R1_001.fastq.gz"
],
"R2": [
"/MAESTRO/test/tutorial/data/time_data/CLL0_S1_L001_R2_001.fastq.gz"
],
"R3": [
"/MAESTRO/test/tutorial/data/time_data/CLL0_S1_L001_R3_001.fastq.gz"
]
},
"CLL24": {
"R1": [
"/MAESTRO/test/tutorial/data/time_data/CLL24_S1_L001_R1_001.fastq.gz"
],
"R2": [
"/MAESTRO/test/tutorial/data/time_data/CLL24_S1_L001_R2_001.fastq.gz"
],
"R3": [
"/MAESTRO/test/tutorial/data/time_data/CLL24_S1_L001_R3_001.fastq.gz"
]
},
"CLL8": {
"R1": [
"/MAESTRO/test/tutorial/data/time_data/CLL8_S1_L001_R1_001.fastq.gz"
],
"R2": [
"/MAESTRO/test/tutorial/data/time_data/CLL8_S1_L001_R2_001.fastq.gz"
],
"R3": [
"/MAESTRO/test/tutorial/data/time_data/CLL8_S1_L001_R3_001.fastq.gz"
]
},
"PBMC0": {
"R1": [
"/MAESTRO/test/tutorial/data/time_data/PBMC0_S1_L001_R1_001.fastq.gz"
],
"R2": [
"/MAESTRO/test/tutorial/data/time_data/PBMC0_S1_L001_R2_001.fastq.gz"
],
"R3": [
"/MAESTRO/test/tutorial/data/time_data/PBMC0_S1_L001_R3_001.fastq.gz"
]
},
"PBMC24": {
"R1": [
"/MAESTRO/test/tutorial/data/time_data/PBMC24_S1_L001_R1_001.fastq.gz"
],
"R2": [
"/MAESTRO/test/tutorial/data/time_data/PBMC24_S1_L001_R2_001.fastq.gz"
],
"R3": [
"/MAESTRO/test/tutorial/data/time_data/PBMC24_S1_L001_R3_001.fastq.gz"
]
},
"PBMC8": {
"R1": [
"/MAESTRO/test/tutorial/data/time_data/PBMC8_S1_L001_R1_001.fastq.gz"
],
"R2": [
"/MAESTRO/test/tutorial/data/time_data/PBMC8_S1_L001_R2_001.fastq.gz"
],
"R3": [
"/MAESTRO/test/tutorial/data/time_data/PBMC8_S1_L001_R3_001.fastq.gz"
]
}
}Step 3. Run snakemake pipeline
Before running the workflow, please check the config.yaml and see if it is configured correctly. Once configured, users can use snakemake to run the workflow.
$ snakemake -np
$ nohup snakemake --cores 32 > multi-scatac-chromap &
##Instead of using nohup, submitting $ snakemake -j 16 by slurm is highly recommended.Step4. Final Outputs
$ ls Result
Analysis Benchmark Log Mapping QC Report
$ ls Result/Analysis
Batch CLL0 CLL24 CLL8 Cluster PBMC0 PBMC24 PBMC8- Analysis/: The Analysis directory contains a Seurat R object, as well as clustering result, cell type annotation result and driver transcription factor identification result. Specificly for the multi-sample scATAC-seq, we process a pseudo-bulk peak calling within each cluster and output the fragment files, narrow peak files, and bigwig files.
- Batch/: All samples merged Results.
- Cluster/: Fragment and bigwig files for each sample within each cluster.
- Benchmark/: The Benchmark directory stores benchmark files for all rules in Snakefile, each of which contains a tab-separated table of run times and memory usage in MiB.
- Log/: The Log directory contains the log files generated in the pipeline analysis.
- Mapping/: The Mapping directory contains all the mapping results generated by chromap or minimap2.
- QC/: The QC directory contains quality control analysis results of scATAC-seq data, including the barcode filtering table outprefix_scATAC_validcells.txt, filtered count matrix outprefix_filtered_peak_count.h5 and other QC results. The filtered count matrix is generated according to singlecell.txt and the parameters like –count-cutoff and –frip-cutoff that users provide.
- Report/: The html file in Report directory summarizes all the results in an HTML based document.
sessionInfo()R version 4.1.0 (2021-05-18)
Platform: x86_64-apple-darwin17.0 (64-bit)
Running under: macOS Big Sur 10.16
Matrix products: default
BLAS: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRblas.dylib
LAPACK: /Library/Frameworks/R.framework/Versions/4.1/Resources/lib/libRlapack.dylib
locale:
[1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] workflowr_1.6.2
loaded via a namespace (and not attached):
[1] Rcpp_1.0.6 whisker_0.4 knitr_1.33 magrittr_2.0.1
[5] R6_2.5.0 rlang_0.4.11 fansi_0.5.0 highr_0.9
[9] stringr_1.4.0 tools_4.1.0 xfun_0.24 utf8_1.2.1
[13] git2r_0.28.0 jquerylib_0.1.4 htmltools_0.5.1.1 ellipsis_0.3.2
[17] rprojroot_2.0.2 yaml_2.2.1 digest_0.6.27 tibble_3.1.2
[21] lifecycle_1.0.0 crayon_1.4.1 later_1.2.0 sass_0.4.0
[25] vctrs_0.3.8 promises_1.2.0.1 fs_1.5.0 glue_1.4.2
[29] evaluate_0.14 rmarkdown_2.9 stringi_1.6.2 bslib_0.2.5.1
[33] compiler_4.1.0 pillar_1.6.1 jsonlite_1.7.2 httpuv_1.6.1
[37] pkgconfig_2.0.3